
New Drug Screening Funnel Could Speed New Therapies for Gaucher Disease, Parkinson’s
October 11, 2024
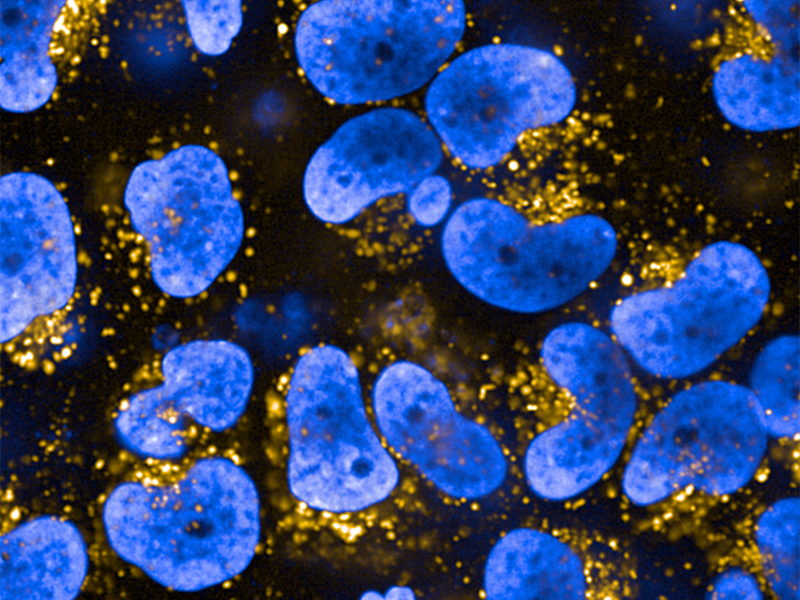
In this image, the pharmacological chaperone NCGC326 rescues GCase enzymatic activity in lysosomes (yellow) of cells with Gaucher mutations (blue). (NCATS)
Scientists could soon streamline their search for new drugs to treat people with Gaucher disease and Parkinson’s disease, thanks to an innovative three-step drug testing process developed by NCATS scientists and their colleagues.
A research team from NCATS, the National Human Genome Research Institute (NHGRI), Simon Fraser University (SFU) and the Roche Innovation Center Basel created a screening approach that can sift thousands of drug compounds. Their design rapidly revealed drugs that can rescue and help restore defective versions of an enzyme that’s a common contributor to both Gaucher and Parkinson’s diseases. Their findings appear in the journal Proceedings of the National Academy of Sciences.
“Developing a platform that enables the discovery and characterization of potential treatments for Gaucher and Parkinson’s is the result of more than a decade of collaborative work,” said Mark Henderson, Ph.D., a co-corresponding author on the study. Henderson is a biology group leader in the NCATS Division of Preclinical Innovation. His NCATS coauthors include study co-lead author Darian Williams, Ph.D., Abhijeet Kapoor, Ph.D., and Juan Marugan, Ph.D.
The enzyme is beta-glucocerebrosidase (GCase). GCase works to help break down glycolipids. Glycolipids provide energy and play a role in maintaining cell membranes. Defective GCase has been linked to Parkinson’s disease, where it plays a role in the damage and death of nerve cells that help the brain control movement.
In Gaucher disease — a rare, inherited disease — a mutant gene leads to faulty GCase. Misfolded GCase can sap cells’ energy production and snarl their ability to break down lipids, proteins and other molecules in a tiny cellular compartment called the lysosome. When this disposal system is impaired, glycolipids build up in such organs as the liver, spleen, bone and — at times — the brain.
Two routes are available for treating Gaucher disease: enzyme replacement and enzyme enhancement.
Enzyme replacement therapy is the standard of care for Gaucher disease. Currently, enzyme replacement therapy doesn’t modify existing faulty GCase, however. It also can’t penetrate the blood–brain barrier to treat neurologic symptoms.
Unlike enzyme replacement therapy, enzyme enhancement therapy (EET) salvages faulty but still partially functional GCase from a cell’s protein disposal system. EET includes two types of molecules that can potentially cross the blood-brain barrier. Proteostasis regulators change how cells handle faulty GCase. Pharmacological chaperones stabilize faulty GCase.
Three Steps to Promising Treatments
The research team designed a three-step screening funnel to find effective EET drug candidates for Gaucher disease. Their process uses quantitative high-throughput screening (qHTS) in all three steps to rapidly narrow the search for promising treatments. The qHTS approach lets scientists test many compounds at different doses all at once.
Before the team could assess any effects of a treatment on faulty GCase, they needed a way to measure GCase’s levels and activity. To do that, they added a tiny piece of genetic code into the mutated GBA1 gene. GBA1 serves as the blueprint for faulty GCase. That new bit of code created a tagged molecule that allowed researchers to easily measure the mutated GCase levels in response to drug treatment.
In the screening funnel’s first stage, the researchers tested the effects of 10,779 compounds on cells with the specially tagged mutated GCase. They assessed each of the compounds at seven different doses. After the initial screening and a follow-up test, 140 of the compounds were judged to be potential hits. Two EET compounds known to work on faulty GCase also cleared the first stage as potential therapies. Their effectiveness showed that the cells with specially tagged mutated GCase mimicked how real cells with faulty GCase work.
In the second stage, the research team created two more high-throughput tests using the cells with faulty GCase to assess some of the successful compounds from the first stage. One test visualized how well GCase was shepherded to the lysosome. The second test gauged GCase levels in the lysosome.
The screening funnel’s third stage paired some of the most promising proteostasis regulators from the first two stages with an effective pharmacological chaperone. The team found a regulator–chaperone combination that completely reversed lipid buildup in the lysosomes of cells with faulty GCase.
A Promising Model for Many Lysosomal Storage Disorders
Since designing their three-step process, the research team has gained a better understanding of how and where its promising chaperone molecules bind to GCase. That knowledge has helped narrow subsequent searches to compounds targeting that binding site.
“This study demonstrates how the focus on a rare disorder like Gaucher disease can lead to insights and therapeutic strategies relevant for far more common disorders like Parkinson’s, a neurodegenerative disease afflicting millions of individuals worldwide,” said Ellen Sidransky, M.D., a co-corresponding author on the study and branch chief at NHGRI. “Our progress would not have been possible without the critical tools and expertise NCATS, NHGRI, SFU, and Roche each brought to this collaboration. It is truly a team effort.”
Sidransky has spent decades investigating Gaucher disease. She won the 2024 Breakthrough Prize in Life Sciences for discovering that mistakes in GBA1, the Gaucher gene, are the most common known genetic risk factor for developing Parkinson’s disease.
The study’s three-stage screening approach can be used for conditions beyond Gaucher and Parkinson’s diseases.
“Theoretically, any lysosomal storage disorder caused by a misfolded protein could be targeted using the approaches that we describe,” said study coauthor Williams, a postdoctoral fellow in the NCATS Division of Preclinical Innovation.